
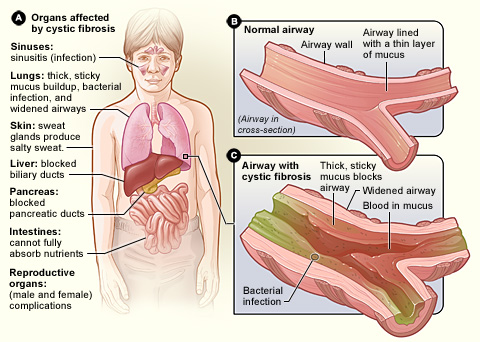
Cystic fibrosis
Cystic fibrosis is a progressive, genetic disease that causes persistent lung infections and limits the ability to breathe over time. In people with CF, mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene cause the CFTR protein to become dysfunctional.
Cystic fibrosis is an inherited disorder that causes severe damage to the lungs, digestive system and other organs in the body.
Cystic fibrosis is a hereditary disease that affects the lungs and digestive system. The body produces thick and sticky mucus that can clog the lungs and obstruct the pancreas.
Cystic fibrosis (CF) can be life-threatening, and people with the condition tend to have a shorter-than-normal life span.
Sixty years ago, many children with CF died before reaching elementary school age. However, advances in treatment mean that people with CF often live into their 30s, 40s, and beyond.
There is currently no cure for CF. It affects some 30,000 people in the United States with around 1,000 new cases diagnosed each year.Of these new diagnoses, 75 percent are made in children under the age of 2 years.
Cystic fibrosis affects the cells that produce mucus, sweat and digestive juices. These secreted fluids are normally thin and slippery. But in people with cystic fibrosis, a defective gene causes the secretions to become sticky and thick. Instead of acting as a lubricant, the secretions plug up tubes, ducts and passageways,especially in the lungs and pancreas.
Although cystic fibrosis requires daily care, people with the condition are usually able to attend school and work, and often have a better quality of life than people with cystic fibrosis had in previous decades. Improvements in screening and treatments mean people with cystic fibrosis now may live into their mid- to late 30s, on average, and some are living into their 40s and 50s.
Table of Contents
Anatomy
Bronchioles – Obstructed secondary to the thickened mucus adhering to the walls of the airways.
Small intestine – distal obstruction (meconium ileus)
Pancreatic duct – Obstruction
Bile ducts – Obstruction
Male reproductive organs – absent or obstructed vas deferens
Female reproductive organs – cervical canal obstructed by thickened mucus
Musculoskeletal system:
– Inspiratory muscle atrophy
- Weakness/atrophy in anti- gravity muscles such as the gastrocnemius
- Kyphosis of the spine resulting in neck and back pain
Mechanism of Injury
Cystic fibrosis (CF) is the most frequent cause of suppurative lung disease in the younger Caucasian population. A depleted volume of the airway surface liquid (ASL) layer in the respiratory system leads to abnormal mucociliary clearance. A chronic cycle of infection and inflammation results in progressive suppurative bronchiectasis and lung damage.
Cystic Fibrosis is an inherited disease of the mucus and sweat glands (exocrine glands) affecting mostly the lungs, liver, pancreas and intestines. It causes damage to lung tissue, inflammation, and acute susceptibility to bacterial infections. There is an abnormal gene, called Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), which results in the production of thick, sticky mucus which blocks the airways in the lungs resulting in frequent lung infection. The thick mucus also blocks the ducts in the pancreas which in turn blocks digestive pancreatic enzymes from reaching the small intestine and performing their normal function.
The degree of mutation of the CFTR gene determines the type of complications that will arise and these will differ from person to person. The mutated or defected protein attaches to the outer membrane of cells in sweat glands, lungs, pancreas and other affected organs. This protein spans over the membrane which acts as a channel connecting the inner part of the cell cytoplasm to the surrounding fluid. This channel is important in our airways because it controls the movement of chloride from the inside to the outside of the cell. Chloride moves from the sweat into the cytoplasm but due to the mutation of the CFTR protein, the chloride is trapped inside the cells of the airway and outside the skin. Chloride is a negatively charged ion and sodium is a positively charged ion, so this leads to an electrical attraction, which results in the formation of salt. In CF patients a high amount of salt is lost in sweat, thus forming the basis of the sweat test.
There are 3 theories:
1) The lack of chloride exodus through the CFTR protein leads to the accumulation and an increase in the viscosity of the nutrient-rich mucus in the lungs leading to bacteria hiding from the body’s immune system.
2) There is a paradoxical increase in the sodium and chloride uptake due to the CFTR protein defect. This cause an increase in the water reabsorption which leads to thick and dehydrated mucus.
3) This theory focuses on abnormal chloride movement out of the cell which leads to dehydrated mucus, pancreas, secretion, biliary secretion etc
These theories propose that the major damage in patients with CF is due to the blockage of narrow passages of the affected organs due to thick secretions. The blockage within the lung airways leads to infections because of the accumulation of the enzymes and blockages of the passages.
Facts on cystic fibrosis
Cystic fibrosis (CF) involves the production of mucus that is much thicker and more sticky than usual.
It mainly affects the lungs and digestive system.
CF is a hereditary condition that occurs in a child when both parents have the defective gene.
All newborns in the U.S. are screened for CF.
There is no cure, but good nutrition and taking steps to thin mucus and improve mucus expectoration can help.
Symptoms of Cystic fibrosis
Screening of newborns for cystic fibrosis is now performed in every state in the United States. As a result, the condition can be diagnosed within the first month of life, before symptoms develop. For people born before newborn screening was performed, it’s important to be aware of the signs and symptoms of cystic fibrosis.
Cystic fibrosis signs and symptoms vary, depending on the severity of the disease. Even in the same person, symptoms may worsen or improve as time passes. Some people may not experience symptoms until adolescence or adulthood.
People with cystic fibrosis have a higher than normal level of salt in their sweat. Parents often can taste the salt when they kiss their children. Most of the other signs and symptoms of cystic fibrosis affect the respiratory system and digestive system. However, adults diagnosed with cystic fibrosis are more likely to have atypical symptoms, such as recurring bouts of inflamed pancreas (pancreatitis), infertility and recurring pneumonia.
Respiratory signs and symptoms
The thick and sticky mucus associated with cystic fibrosis clogs the tubes that carry air in and out of your lungs.
This can cause signs and symptoms such as:
A persistent cough that produces thick mucus (sputum)
Wheezing
Breathlessness
Exercise intolerance
Repeated lung infections
Inflamed nasal passages or a stuffy nose
Digestive signs and symptoms
The thick mucus can also block tubes that carry digestive enzymes from your pancreas to your small intestine. Without these digestive enzymes, your intestines aren’t able to completely absorb the nutrients in the food you eat. The result is often:
Foul-smelling, greasy stools
Poor weight gain and growth
Intestinal blockage, particularly in newborns (meconium ileus)
Severe constipation
Frequent straining while passing stool can cause part of the rectum — the end of the large intestine — to protrude outside the anus (rectal prolapse). When this occurs in children, it may be a sign of cystic fibrosis. Parents should consult a physician knowledgeable about cystic fibrosis. Rectal prolapse in children may sometimes require surgery. Rectal prolapse in children with cystic fibrosis is less common than it was in the past, which may be due to earlier testing, diagnosis and treatment of cystic fibrosis.
Causes of Cystic fibrosis
In cystic fibrosis, a defect (mutation) in a gene changes a protein that regulates the movement of salt in and out of cells. The result is thick, sticky mucus in the respiratory, digestive and reproductive systems, as well as increased salt in sweat.
Many different defects can occur in the gene. The type of gene mutation is associated with the severity of the condition.
Children need to inherit one copy of the gene from each parent in order to have the disease. If children inherit only one copy, they won’t develop cystic fibrosis. However, they will be carriers and possibly pass the gene to their own children.
Risk factors
Family history. Because cystic fibrosis is an inherited disorder, it runs in families.
Race. Although cystic fibrosis occurs in all races, it is most common in white people of Northern European ancestry.
Complications
Respiratory system complications
Damaged airways (bronchiectasis). Cystic fibrosis is one of the leading causes of bronchiectasis, a condition that damages the airways. This makes it harder to move air in and out of the lungs and clear mucus from the airways (bronchial tubes).
Chronic infections. Thick mucus in the lungs and sinuses provides an ideal breeding ground for bacteria and fungi. People with cystic fibrosis may often have sinus infections, bronchitis or pneumonia.
Growths in the nose (nasal polyps). Because the lining inside the nose is inflamed and swollen, it can develop soft, fleshy growths (polyps).
Coughing up blood (hemoptysis). Over time, cystic fibrosis can cause thinning of the airway walls. As a result, teenagers and adults with cystic fibrosis may cough up blood.
Pneumothorax. This condition, in which air collects in the space that separates the lungs from the chest wall, also is more common in older people with cystic fibrosis. Pneumothorax can cause chest pain and breathlessness.
Respiratory failure. Over time, cystic fibrosis can damage lung tissue so badly that it no longer works. Lung function usually worsens gradually, and it eventually can become life-threatening.
Acute exacerbations. People with cystic fibrosis may experience worsening of their respiratory symptoms, such as coughing and shortness of breath, for several days to weeks. This is called an acute exacerbation and requires treatment in the hospital.
Digestive system complications
Nutritional deficiencies. Thick mucus can block the tubes that carry digestive enzymes from your pancreas to your intestines. Without these enzymes, your body can’t absorb protein, fats or fat-soluble vitamins.
Diabetes. The pancreas produces insulin, which your body needs to use sugar. Cystic fibrosis increases the risk of diabetes. Around 30 percent of people with cystic fibrosis develop diabetes by age 30.
Blocked bile duct. The tube that carries bile from your liver and gallbladder to your small intestine may become blocked and inflamed, leading to liver problems and sometimes gallstones.
Intestinal obstruction. Intestinal obstruction can happen to people with cystic fibrosis at all ages. Children and adults with cystic fibrosis are more likely than are infants to develop intussusception, a condition in which a section of the intestines folds in on itself like an accordion.
Distal intestinal obstruction syndrome (DIOS). DIOS is partial or complete obstruction where the small intestine meets the large intestine.
Reproductive system complications
Almost all men with cystic fibrosis are infertile because the tube that connects the testes and prostate gland (vas deferens) is either blocked with mucus or missing entirely. Certain fertility treatments and surgical procedures sometimes make it possible for men with cystic fibrosis to become biological fathers.
Although women with cystic fibrosis may be less fertile than other women, it’s possible for them to conceive and to have successful pregnancies. Still, pregnancy can worsen the signs and symptoms of cystic fibrosis.
Other complications
Thinning of the bones (osteoporosis). People with cystic fibrosis are at higher risk of developing a dangerous thinning of bones.
Electrolyte imbalances and dehydration. Because people with cystic fibrosis have saltier sweat, the balance of minerals in their blood may be upset. Signs and symptoms include increased heart rate, fatigue, weakness and low blood pressure.
Prevention
If you or your partner has close relatives with cystic fibrosis, you both may want to undergo genetic testing before having children. The test, which is performed in a lab on a sample of blood, can help determine your risk of having a child with cystic fibrosis.
If you’re already pregnant and the genetic test shows that your baby may be at risk of cystic fibrosis, your doctor can conduct additional tests on your developing child.
Genetic testing isn’t for everyone. Before you decide to be tested, you should talk to a genetic counselor about the psychological impact the test results might carry.
Diagnosis
All newborns in the U.S. are screened for CF by testing a small blood sample or samples. This can indicate that a baby might have a health condition and require further investigation.
CF is usually diagnosed through a sweat test. Sweat is collected and the amount of chloride, a component of salt in the sweat, is measured. A high level of chloride is an indication of CF.
Genetic tests can also be carried out by analyzing cheek cells or a blood sample. These tests are mainly used to find out if a person carries the CF gene, but they can also be used to confirm a CF diagnosis following an unclear sweat test result.
There are over 1,700 known mutations of the CF gene. As a result, most genetic tests for the condition only screen for the most common mutations.
Seventy-five percent of people with CF are diagnosed by the age of 2 years.
Physiotherapy Treatment
There is currently no cure for CF. Treatment can manage the symptoms of the disease, however, and improve quality of life. Symptoms can vary and treatment plans will be individualized.
Airway clearance
It is crucial for people with CF to get rid of mucus from their lungs to allow clear breathing and minimize lung infections.
Airway clearance techniques (ACT) can help people with CF to loosen and get rid of mucus from their lungs.
An example of ACT would be postural drainage and percussion. A therapist claps the patient’s chest and back while they sit, stand, or lie in a position that should help to free up mucus.
Inhaled medication is effective at reaching the airways and commonly used. The medication can be given by aerosol or as a metered dose inhaler. These medications can thin mucus, kill bacteria, and mobilize mucus to improve airway clearance.
Antibiotics are an important part of regular care. These can be taken orally, intravenously, or through inhalation.
Other drugs, such as ibuprofen and azithromycin, have been found to preserve and improve lung function, and are now considered to be a part of standard therapy for people with CF.
People with CF can also help reduce their risk of lung infection by taking the following steps:
washing the hands frequentlygetting a flu shot every yearnot smoking and avoiding second-hand smoke avoiding unnecessary contact with people who have colds or other contagious illnessesOther forms of treatment.
There are alternative methods of managing CF that do not involve the airways.
Implanted devices can allow long-term access to the bloodstream for the frequent and regular administration of drugs. They can make management of a chronic condition like CF more efficient and less intrusive.
CF transmembrane conductance regulator (CFTR) modulators are newer medications that target the faulty CF-causing gene. They allow for proper flow of salt and fluids on the surface of the lungs, thinning the thick mucus that people with CF usually have built up in their lungs.
Two CFTR modulator brands are currently approved by the Food and Drug Administration (FDA). These are Kalydeco and Orkambi. They are prescribed for children with 10 different mutations of the CF-causing gene.
Kalydeco may be prescribed from the age of 2 years onwards, and Orkambi at 6 years.
Nutritional therapy for digestive symptoms
As CF can affect digestive function and nutrient absorption, people with CF should discuss their diet with their doctor. A nutritionist or dietitian may help with the management of digestive symptoms.
A different kind of diet or additional supplements, such as pancreatic enzyme supplements, salt, or vitamins, may be needed to balance the absorption of nutrients.
CF can lead to impaired growth. A high-calorie, high-fat diet is essential for normal growth and development in children with CF. It can help adults to maintain optimal health.
Good nutrition is vital, as individuals with CF need to maintain a robust defense against an increased risk of lung infection.
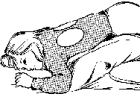
Manual chest physical therapy (CPT) is the traditional airway clearance technique for cystic fibrosis (CF) patients. The removal of obstructive, thick sticky secretions is important in preventing infections and helps ease breathing. Manual chest physiotherapy consists of two parts:
Postural drainage
Percussion and vibration.
Postural drainage pertains to placing the body in a position which allows the mucus to drain from the smaller airways into the main airway with gravity. Two positions are in an upright sitting position and four positions are with the head tilted below the lungs. To obtain the head-down positions, the use of a pillow, a bean bag chair or couch cushions works well.
Percussion and Vibration
Percussion and vibration help loosen and mobilize secretions. Percussion is a repetitive tapping on the designated position and can be done with palm cups (of varying sizes), by hand or with a manual percussor.
If you choose to use your hands, the palm of your hand should be cupped to provide a pocket of air that cushions the percussion. Manual percussors have padding and velcro to allow self-use. Vibration is done at the end of the position. After a big breath in, vibrate when the air is slowly blown out, for three big breaths.
Six Manual Chest Physiotherapy Positions


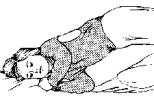
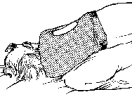

Recommendations
Each position requires proper placement for percussion and should be done for three to five minutes
Nothing should be placed in the mouth (pacifiers, food, etc.) while doing manual chest physiotherapy
A strong cough is encouraged after each position to remove the mucus
It is recommended that manual chest physiotherapy be done before meals and bedtime. Your doctor will determine how often manual chest physiotherapy is needed
Airway clearance is a very important part of the daily cares in cystic fibrosis, and we recommend this a minimum of once per day for all patients (even those without a cough)
As infants grow it may become more challenging for them to cooperate with manual chest physiotherapy. You may find that it helps to make therapy time a special time. This can be done with a toy, watching a video, or a sticker reward. Keep these times special by only giving them with cooperation of therapy and not allowing them to be used any other time.
The main aim of physiotherapy is to remove excessive mucoid secretions in order to improve ventilation. A person suffering from CF will require intensive chest physiotherapy. This will include the following:
Vigorous massage to help loosen the sticky mucus
Postural drainage: gravity assisted positions to help drainage of secretions and also helps to increase the air movement or ventialtion to different parts of the lungs .
Percussions: This technique is also known as chest clapping, and is used to help loosen secretions. To perform this technique a cupped hand is used to clap the chest firmly and rhythmically .
Shaking and Vibrations: This technique consists of several short rhythmical squeezes to the chest while exhaling to mobilze secretions .
Regular assessment and monitoring is necessary during physiotherapy treatment as the patient may require supplemental oxygen, especially in advanced cystic fibrosis.
Other techniques include:
Positioning: lying on either side, front or back and sitting .
Active cycle of breathing (ACBT): This technique consist of Breathing Control (BC), Thoracic Expansion Exercises (TEE), and Forced Expiration Technique (FET)
Breathing Control (BC): Relaxed diaphragmatic breathing which allows for rest and helps avoid any tightening of airways, which can make it difficult to clear secretions .
Thoracic expansion exercises (TEE): Deep breathing exercises that help the lungs to expad more effectively and allow air to get behind secretions so that they can be “pushed” up the airways towards the mouth. The breaths should be slow and deep with a pause at the end of inspiration then followed by relaxed quiet expiration .
Forced Expiration Technique (FET): This consists of huffing or a sigh, to help move secretions from the smaller to the larger airways from where they can be cleared more easily.
Positive expiratory pressure (PEP): A technique that helps to open up airways and get air behind secretions to help move them higher up the airway. The PEP device (mask or mouthpiece) gives a small degree of resistance to the breath out and this resistance splints open the airways. Bubble PEP is used in young children and involves the child blowing through a straw placed in a bottle of water and soap to produce bubbles.
Oscillating PEP: These devices combine vibrations of airways with PEP and there are several different types. The Flutter is a small pipe shaped device that interrupts the flow of air and gives an intermittent back pressure to the airways as well as causing them to vibrate. The Cornet and the Acapella are two other oscillating PEP devices that function similarily to the Flutter device .
Gentle huffing
Exercise advice – doing active exercises such as running, swimming, dancing etc.
Parents of a child with CF are taught by hospital staff on how to manage their child. Older children and adults can be taught to do this for themselves. Advice will include all of the techniques above. It is also important to educate the family about the condition and to provide counselling where necessary.
Exercise
The short and long term benefits of regular physical activity are well established and the importance of regular physical exercise as an essential part of the physiotherapy regimen is recognized. The effect of inspiratory muscle training in CF however remains unclear.
Benefits of Exercise for the CF patient include :
enhanced fitness levels
increased sputum clearance
delay the onset of dyspnea
delay declines in pulmonary function
prevent decrease in bone density
enhance cellular immune response
increase feelings of well-being
may improve self-image, self-confidence, and quality of life
Exercise in the Literature
Recent literature suggests that exercise produces similar benefits as the traditional airway clearance techniques and exercise should be encouraged over these techniques when clinically appropriate for the patient. Exercise is beneficial in that evidence suggests that exercise may even reverse some of the disease related changes in ion transport resulting in a decrease of the thickened mucus and therefore leading to improved pulmonary functioning.
In a clinical trial by Elbasan et al. clinically stable children with cystic fibrosis (FEV1<35%) were taken through a training program that consisted of active cycles of breathing techniques and aerobic exercise training on the treadmill using 74-80% of the maximum heart rate for 30 minutes. This was performed three times a week for six weeks. The active cycles of breathing techniques were taught along with posture and breathing to be used as a home exercise program. The study concluded that active cycles of breathing techniques should be included with aerobic exercise to improve physical fitness including thoracic mobility, muscle strength and endurance, flexibility, and speed.
A systematic review of randomized clinical trials by Nancy van Doorn included four out of 599 RCT’s that met the inclusion criteria. Exercise intervention’s included aerobic training, resistance training, anaerobic training and strength training. Strength training was strongly correlated with strength gains which is important to prevent deconditioning in this patient population. Results also suggested that peak VO2 could be improved from short-term aerobic training, which is important for survival rate in these patients. Anaerobic fitness also demonstrated improvement. This systematic review concludes that children with CF can improve pulmonary function through either aerobic or resistance training.
A recent trial study by Reix et al. sought to answer the questions: “1. Can session of exercise with incorporated maneuvers substitute for a session of breathing techniques for airway clearance in children with cystic fibrosis? and 2. Are children with cystic fibrosis as co-operative and satisfied with the exercise regimen as with the breathing techniques?” Using a randomized, crossover design “each participant underwent two 20 minute airway clearance interventions on two scheduled clinic days: One involving 3 bouts of various whole-body exercise modalities each followed by independent expiratory maneuvers , and the other involving breathing control, thoracic expansions with manual expiratory compressions, and the forced expiratory technique.” Although the conclusions of this study are very limited by design, they concluded that “a session of various whole-body exercises interspersed with independent expiratory maneuvers could be an acceptable substitute for breathing techniques” and that it may be preferred by children with cystic fibrosis due to the ability to modify the exercise bouts in order to keep it new and challenging.
A study by Paranjape et al. looked at the effects of a home exercise program for children with cystic fibrosis hypothesizing that increasing their activity would lead to improved exercise capacity, lung function, nutritional status, and quality of life. Their outcome measures included the Modified Shuttle Walk Test, BMI, FEV1, Habitual Activity Estimation Scale, and 5 exercise-related domains of the Revised CF Quality of Life Questionnaire. Measures were taken before and after the intervention which included a 2-month, individualized home exercise program. All measurements showed an increase or improvement except for the HAES on weekday activity but only the MSWT and CFQ-R body image perception domain were statistically significant. The conclusion of this study is that individualized, home exercise programs are beneficial for children with cystic fibrosis.
End- of-life care
The patient and the family need more sensitive discussions about prognosis and preferences for care throughout the the course of the disease. Most people facing end of life with cystic fibrosis are older adolscents or young adults and should be informed of the choices they have about their illnesses. Often discussions about transplantation are done to give the patients the chance to weigh the merits of longer survival with a transplant against the detoriorating illness without a transplant. Palliative care such as sedation should be given to ensure peaceful dying, when it is appropriate.

8 Comments