Huntington Disease
Table of Contents
Introduction
Huntington’s disease (HD) is a genetic neurodegenerative disorder that affects the central nervous system. It is named after George Huntington, the physician who first described the condition in 1872. HD is characterized by the progressive breakdown of nerve cells in certain regions of the brain, leading to various physical, cognitive, and psychiatric symptoms.
What is the main Huntington’s disease (HD)?
- Huntington’s disease (HD) is mainly a genetic disease passed from parent to child. . many Symptoms get worse over time. They eventually affect walking, talking, or swallowing. It also affects psychological areas such as mood swings or memory problems.
- HD is caused by a mutation in the huntingtin (HTT) gene, which produces a protein called huntingtin. In individuals with HD, the gene contains an abnormal repetition of the CAG nucleotide sequence. The sooner the illness develops and the more severe it becomes, the more CAG repeats there are.
- Symptoms of Huntington’s disease typically appear between the ages of 30 and 50, although there are cases of juvenile-onset HD that manifest before the age of 20. The hallmark symptom of HD is involuntary jerking and writhing movements, known as chorea
Whom does Huntington’s disease (HD) affect?
This disease can affect anyone, However mostly it is found in people of European descent (having family members who came from Europe). It also a genetic disease means whether your parent has Huntington’s disease. If so you are at risk of a 50% chance of also having the HD disease.
What is juvenile main Huntington’s disease (HD)?
Mostly This disease symptoms start in middle age, However, in a few cases where it starts to appear in childhood, it is called juvenile Huntington’s disease. In Juvenile HD, other symptoms such as seizures or stiffness are also associated. JHD is mostly occurred due to inheriting the main disease from their fathers.
How common is the main Huntington’s disease (HD)?
It is a rare disease affecting about 30,000 people in America. In North America it is found in 5 people per 10 lakh while juvenile HD is also rare, found in 5 to 10% of people of total HD cases.
How is the main Huntington’s disease (HD) inherited?
- You need to have a basic understanding of genetics, which is the study of how physical characteristics are transmitted down from one generation to the next, in order to fully understand how Huntington’s disease is inherited. Your body has DNA (deoxyribonucleic acid) in every cell. DNA is your body is instruction manual. It offers the knowledge required to rebuild or heal cells. Your DNA dictates everything from your hair color or height to how your organs function.
- The major “chapters” in the DNA instruction book are analogous to the genes. Here is how the main genes affect Huntington’s disease:
- Your body is given directions on how to create the primary huntingtin protein by the huntingtin gene (HTT or HD gene). Each parent contributes just one HTT gene to you. If you have the main Huntington’s disease, one of your parents passed on an HTT gene with a mutation (like a misprint in a book). It tells your body to create a not used long protein.
- Researchers believe this long protein damages or kills brain cells. The primary symptoms will ultimately manifest in everyone who inherits the gene. The exact age when many symptoms appear varies. Each successive generation tends to experience HD symptoms earlier than the one before it.
How does main Huntington’s disease (HD) affect the brain?
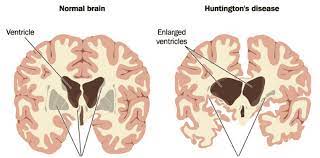
- Huntington’s disease arises from the destruction of neurons (brain cells) by misshaped proteins. They often start by attacking the basal ganglia, a region of the brain that controls the majority of the motions in your body. The cortex, or outer layer of the brain, is also affected by the condition. This area of the brain aids with memory, thought, and decision-making.
What are the main symptoms of Huntington’s disease (HD)?
- A person with HD has both physical and mental effects. Small physical issues, like clumsiness and losing your balance, might develop into larger ones over time. Huntington’s disease patients may have chorea, or uncontrolled movements.
- Emotional changes like main mood swings, depression, or irritability.
- Problems with memory, focus, or multitasking.
- Slowed movements or speech.
- Slurred speech.
- Reduce hand coordination, such as being unable to hold a pencil.
- Difficulty swallowing.
What is the main Huntington’s disease chorea?
- One of the earliest physical signs of HD is chorea, which includes unintentional jerks and twisting motions. Chorea usually affects the hands, fingers, or face muscles first. Later, it also makes your arms, legs, or torso move uncontrollably. Chorea can make speaking, eating, or walking more difficult. It may also affect your main ability to perform all daily activities such as driving.
Can main Huntington’s disease (HD) cause dementia?
- In its later stages, Huntington’s disease can cause dementia. Memory loss or personality changes result from brain function loss. The effects on your primary brain are different earlier in the disease. You might have problems multitasking and doing something that involves multiple steps. It could also be hard to plan and problem-solve soon after the onset of Huntington’s disease.
How is main Huntington’s disease (HD) diagnosed?
- A primary physical examination will be conducted by a neurologist (a physician with expertise in the brain and nerves). They will look for twitches or jerking as well as problems with your balance, reflexes, or coordination. If there is anyone else in your family who has the condition, your primary neurologist will also want to know.
- You will undergo testing to confirm an HD diagnosis and to rule out other major illnesses that commonly produce similar signs and symptoms. Tests include:
- Blood test.
- Genetic testing.
- Imaging tests such as magnetic resonance imaging (MRI) and main computed tomography (CT) scan.
What is main genetic testing like?
- Your healthcare provider draws your blood or sends it off to a laboratory to look at your DNA. The primary test confirms if you carry a mutation in the primary HTT gene. The major topics you’ll cover with a genetic counsellor (someone who specializes in genetic testing) are the testing process and results.
- Occasionally, genetic testing does not provide a clear answer. Your primary HTT gene may be in the centre, producing longer-than-average proteins but not offering a clear solution. In that case, main imaging tests may help. They can reveal if your brain has undergone significant HD-related alterations. Your doctor could also inquire as to whether other members of your family can undergo genetic testing with you.
Can you find out if you have Huntington’s disease (HD) before many symptoms appear?
- If one of your parents and siblings has HD, your risk of having it is high. If you have a gene mutation, predictive genetic testing, which looks for genetic disorders before many symptoms appear, can determine this. People have different responses to learning about a disease they all get someday. Making family or financial planning may be aided by being aware of the gene. However, it could also be emotionally challenging, particularly because you can’t stop the condition. It is crucial to talk about the primary tests with your genetic counsellor to determine if learning is the best course of action for you.
Differential diagnosis
- About 99% of HD diagnoses based on the typical symptoms or a family history of the disease are confirmed by genetic testing to have the expanded trinucleotide repeat that causes HD. Many of the remaining are called HD-like (HDL) syndromes. Many HDL disorders have unknown origins, however those that do have explanations are brought on by mutations in the prion protein gene (HDL1), the TATA box-binding protein gene (SCA17, also known as HDL4), the junctophilin 3 gene (HDL2), or both.
- HDL3—a recessively inherited unknown gene that is only found in two families or is poorly understood. Atrophy is another autosomal dominant condition that may be mistakenly identified as HD. Additionally, certain autosomal recessive diseases mirror the majority of sporadic HD cases. These include chorea acanthocytosis or pantothenate kinase-associated neurodegeneration. The McLeod syndrome is one X-linked illness of this primary kind.
Treatment
Is there a cure for main Huntington’s disease (HD)?
- There is no cure for Huntington’s disease. However, medicines that reduce unusual huntingtin protein are now the subject of clinical studies (testing on humans) to determine their safety and efficacy.
How is main Huntington’s disease (HD) treated?
- Because HD affects you in various ways — physical, emotional, or mental — you may need several types of treatment. Together, physical therapy, counselling, and/or medicine may help you feel better. A multi-disciplinary approach utilizing a neurologist, psychiatrist, genetic counseling, physical therapist, occupational therapy, speech therapy, or other specialized fields can formulate a plan and address a patient’s individual needs.
- To control chorea, doctors more commonly prescribe:
- Tetrabenazine (Xenazine®).
- Deutetrabenazine (Austedo®).
- Haloperidol (Haldol®).
- Your doctor may suggest the following to treat problems that are largely emotional:
- Antidepressants: Drugs that relieve depression include fluoxetine (Prozac®, Sarafem®) or sertraline (Zoloft®).
- Antipsychotic drugs: To reduce angry outbursts, agitation, or hallucinations, your doctor may recommend drugs such as risperidone (Risperdal®) and olanzapine (Zyprexa®).
- Mood-stabilizing drugs: Medications like lithium (Eskalith®) decrease anxiety or prevent severe mood swings.
Can you prevent main Huntington’s disease (HD)?
- A HTT gene mutation leads to Huntington’s disease. You can’t change your genes and prevent the disease from developing. Currently, there isn’t a treatment that can slow and stop the progression of HD.
Prognosis
- The length of the trinucleotide repeats accounts for 60% of the variation of the age of many symptoms onset and their rate of progress. A longer repeat results in an earlier age of onset or a faster progression of many symptoms. The remaining variation is due to environmental factors or other genes that influence the mechanism of the disease. The average HD patient lives 10 to 30 years after the first noticeable symptoms appear. Ten years after the development of obvious symptoms in many systems, juvenile Huntington’s disease patients can expect to live.
- Muscle coordination and, to a lesser extent, behavioral alterations brought on by deteriorating cognitive function are the main causes of life-threatening problems. The largest risk is pneumonia, which causes death in one-third of those with Huntington’s disease. The risk of developing pneumonia increases with a decline in motor coordination, difficulties emptying the lungs, or a higher chance of aspirating food and drink. Heart disease, which accounts for over a quarter of Huntington’s disease fatalities, is the second-highest risk. Suicide is the third leading cause of death, with up to 27% of HD patients attempting suicide and 7.3% actually completing it.
- It is unknown how much behavioral indicators, which indicate a wish to escape the most severe phases of the illness, affect suicidal thoughts. Suicide is the greatest risk of this disease before the diagnosis is made or in the middle stages of the development of the disease. Malnutrition, physical harm from falls, and choking owing to the inability to swallow are some of the other concerns.
Will main Huntington’s disease (HD) kill you?
- Daily tasks become progressively more challenging for people with Huntington’s disease over time. Depending on the individual, it might proceed more quickly or slower. But the average lifespan after most diagnoses is 10 to 30 years. HD itself is not fatal. However, its side effects, including infections like pneumonia and injuries from falls, might cause death.
When I have primary Huntington’s disease (HD), how can I take care of myself?
- As the primary condition advances, there are various actions you may take to maintain the highest quality of life, including:
- Get regular exercise: Research shows exercise helps decrease symptoms.
- Eat a healthy diet: You may need extra main nutrition.
- Drink plenty of water: When swallowing becomes most difficult, you can easily become dehydrated.
- Find a support group: Ask your doctor for mainly community resources where you can connect with others affected by the disease.
- Research care services: At some point, you’ll need a high level of care from either home care services or a nursing home.
- Appoint a reliable advisor: As the illness worsens, you may need to assign money management or critical decision-making to someone else.
- Though you cannot avoid the major form of Huntington’s disease, you can prepare for it.
- Though you cannot prevent the major form of Huntington’s disease, you can prepare for it. many Symptoms take years to worsen. That gives you time to find healthcare providers you trust or get the support you need for the future. Ask a genetic counsellor what you need to know if you or a member of your family has Huntington’s disease.
FAQs
A huntingtin disease protein gene mutation, which leads to HD, is responsible for this condition. The deficiency makes the cytosine, adenine, or guanine (CAG) DNA building components repeat many more times than they should. The majority of people are not at risk for the condition since their Huntington’s disease gene has less than 27 CAG repeats.
Uncontrolled movement of the arms, legs, head, face, or upper torso is a defining feature of many primary Huntington’s disease symptoms.
Additionally, thinking or reasoning abilities such as memory, focus, judgment, or the capacity to plan or organize suffer from Huntington’s disease.
41.3% of the primary patients reported overall pain. The prevalence of primary pain may range from 10% to 75% depending on the research.
The number of individuals who experience discomfort is equivalent to those of other neurodegenerative disorders, including Parkinson’s disease, according to researchers.
advanced stages of Huntington’s condition A person with Huntington’s disease at this stage is unable to work, manage their own finances, take care of themselves, or handle household duties. They may also have mobility issues and spend the most of their time in a chair or bed.
No treatments can alter the course of main Huntington’s disease. But medications can lessen some symptoms of movement or
psychiatric disorders.
Occupational therapy
Handrails at home.
Assistive devices for activities such as bathing or dressing.
Eating or drinking utensils are adapted for people with limited fine motor skills.