MYOTONIA
Table of Contents
INTRODUCTION:
- Individuals with myotonia may have trouble releasing their grip on objects or may have difficulty rising from a seated position. They may walk with a stiff, awkward gait. Myotonia is caused by an abnormality in the muscle membrane and is often associated with inherited neurological disorders.
- Myotonia is commonly seen in individuals with myotonic muscular dystrophy, myotonia congenital, and in people who have one of a group of neurological disorders called the channelopathies, which are inherited diseases that are caused by mutations in the chloride sodium or potassium channels that regulate the muscle membrane. Myotonia may also be triggered by exposure to cold.
- Myotonia is, by definition, the impairment of relaxation of skeletal muscles after voluntary contraction or electrical stimulation. Many etiologies result in myotonia, including dystrophic and non-dystrophic myotonias. Myotonic dystrophies are among the more common muscular dystrophies, while the non-dystrophic myotonias can be quite rare and thus frequently misdiagnosed.
DEFINITION:
- Myotonia is a medical term that refers to a neuromuscular condition in which the relaxation of a muscle is impaired. It can affect any muscle group. The repeated effort will be needed to relax the muscle, although the condition usually improves after the muscles have warmed up.
- Myotonia is a symptom of a small handful of certain neuromuscular disorders characterized by delayed relaxation (prolonged contraction) of the skeletal muscles after voluntary contraction or electrical stimulation.
- Myotonia is the defining symptom of many channelopathies such as myotonia congenita, paramyotonia congenital, and myotonic dystrophy.
- Generally, repeated contraction of the muscle can alleviate the myotonia and relax the muscles thus improving the condition, however, this is not the case in paramyotonia congenital. This phenomenon is known as the “warm-up” reflex and is not to be confused with warming up before exercise, though they may appear similar. Individuals with the disorder may have trouble releasing their grip on objects or may have difficulty rising from a sitting position and a stiff, awkward gait.
- Myotonia can affect all muscle groups; however, the pattern of affected muscles can vary depending on the specific disorder involved.
- People suffering from disorders involving myotonia can have life-threatening reactions to certain anesthetics called anesthesia-induced rhabdomyolysis.
ETIOLOGY:
The presentation of myotonia can result from a diverse array of etiologies. The most common myotonic disorder is myotonic dystrophy type 1, resulting from a trinucleotide repeat on the dystrophia myotonica protein kinase (DMPK) gene that has varying protein consequences depending on the length of the repeat. The next most common disorders are the myotonic channelopathies, led by myotonia congenita in prevalence, which results from defects in electrolyte channels that cause resulting skeletal muscle over excitability.
THERE ARE 3 TYPES:
1.Dystrophic myotonias
-Myotonic dystrophy type 1
-Myotonic dystrophy type 2
2.Non-dystrophic myotonias
-Myotonia congenita
-Paramyotonia congenita
-Sodium channel myotonias
3.Periodic paralyzes
-Hypokalemic periodic paralysis type 1
-Hypokalemic periodic paralysis type 2
-Hyperkalemic periodic paralysis
-Andersen-Tawil syndrome
CAUSES:
1) Myotonic dystrophy:
- Myotonic dystrophy is a disease that affects the muscles and other body systems. It is the most common form of muscular dystrophy that begins in adulthood, usually in a person’s 20s or 30s. This disease is characterized by progressive muscle loss and weakness. Myotonic dystrophy may be further classified into two types, and the two types may affect different muscles. People with myotonic dystrophy usually have prolonged muscle tensing (myotonia) and are not able to relax certain muscles after use. The severity of the disease may vary among affected people, even among members of the same family.
- Myotonic dystrophy is caused by mutations (changes) in the DMPK gene or the CNBP (ZNF9) gene depending on the specific type of myotonic dystrophy. The disease is inherited in an autosomal dominant manner. Myotonic dystrophy may be diagnosed when a healthcare provider observes signs and symptoms of the disease, and the diagnosis may be confirmed with tests of muscle function and genetic testing. Treatment is based on each person’s specific signs and symptoms and may include physical therapy, pain management with medication, and consultation with specialists.
Symptoms:
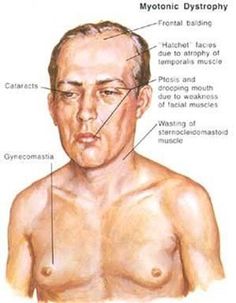
- Signs and symptoms of myotonic dystrophy often begin in a person’s 20s or 30s but can begin at any age. Symptoms often include progressive muscle weakness, stiffness, tightness, and wasting. There are two types of myotonic dystrophy: myotonic dystrophy type 1 and myotonic dystrophy type 2. The symptoms in people with myotonic dystrophy type 2 tend to be milder than in those with type 1, but the symptoms may overlap. People with myotonic dystrophy type 1 typically experience involvement of the legs, hands, neck, and face, while people with myotonic dystrophy type 2 typically experience involvement of the neck, shoulders, elbows, and hips. The severity of symptoms can vary widely among affected people.
- Other signs and symptoms of myotonic dystrophy can include cataracts, type-2 diabetes, and cardiac conduction defects (irregular electrical control of the heartbeat). Some affected men also have hormonal changes that may cause balding or infertility.
- In some cases, babies are born with a variation of myotonic dystrophy type 1 called congenital myotonic dystrophy. Symptoms of congenital myotonic dystrophy are present from birth and include weakness of all muscles, breathing problems, clubfeet, developmental delays, and intellectual disabilities.
Cause:
- Myotonic dystrophy is caused by mutations (changes) in either the DMPK gene (in type 1) or the CNBP (ZNF9) gene (in type 2). The specific kinds of mutations found in both types of myotonic dystrophy are trinucleotide repeat expansions. These types of mutations occur when a piece of DNA is abnormally repeated a number of times, which makes the gene unstable. The mutated gene makes an altered version of messenger RNA (mRNA), which is a copy of the gene that is normally used for protein production. The abnormal mRNA forms clumps inside the cell that interfere with the production of many proteins. These changes prevent cells in muscles and other tissues from functioning normally, leading to the signs and symptoms of myotonic dystrophy.
- The exact functions of the DMPK and CNBP genes are not well understood. DMPK may play a role in communication within cells, specifically in cells of the heart, brain, and skeletal muscles. The CNBP gene gives the body directions to make a protein found mainly in the cells of the heart and skeletal muscles, where it is thought to regulate the activities of other genes.
Inheritance:
- Myotonic dystrophy (DM) is inherited in an autosomal dominant pattern. This means that one copy of the altered gene in each cell of the body is enough to cause symptoms of the disease. We inherit one copy of each gene from our mother and the other from our father. Therefore, when a person with myotonic dystrophy goes on to have children, there is a 50% chance that each child will have myotonic dystrophy.
Diagnosis:
- Myotonic dystrophy is diagnosed by doing a physical exam. A physical exam can identify the typical pattern of muscle wasting and weakness of the jaw and neck muscles and the presence of myotonia. Men may have frontal balding.
- There are several laboratory tests that can be used to clarify the clinical diagnosis of myotonic dystrophy. One test, called electromyography (EMG), involves inserting a small needle into the muscle. The electrical activity of the muscle is studied and usually shows characteristic patterns of myotonic dystrophy.[3] Other laboratory tests may include a muscle biopsy, which can be used to determine if the muscle fibers are weaker than they should be (atrophied), or a blood test to determine if there are elevated levels of certain muscle enzymes.
- The definitive test for myotonic dystrophy is a genetic test. For this test, a blood or saliva sample is analyzed to determine if there are a mutation in the DMPK or CNBP (ZNF9) genes.
- Any person who has myotonic dystrophy can pass the mutation on to his or her children, whether the children are male or female. Myotonic dystrophy type 1 exhibits an unusual genetic pattern called anticipation. Anticipation means the signs and symptoms of a genetic disease begin earlier in life and become more severe as the disease is passed on through generations. Congenital myotonic dystrophy type 1 occurs only when the disease is inherited from the mother.
Treatment:
- There is currently no cure or specific treatment for myotonic dystrophy. Treatment is aimed at managing symptoms of the disease. Routine physical activity appears to help maintain muscle strength and endurance and to control musculoskeletal pain. Canes, braces, walkers, and scooters can help as muscle weakness progresses.
- There are also medications that can lessen pain associated with myotonic dystrophy. Pain management can be achieved through the use of medications prescribed by a doctor.
- Heart problems associated with myotonic dystrophy can be treated through the insertion of a pacemaker, medications, and regular monitoring of cardiac function. Cataracts can be surgically removed. Testosterone replacement therapy may be used to treat infertility in males.
Prognosis:
- The long-term outlook (prognosis) for each person with myotonic dystrophy (including life expectancy) may depend on the type of myotonic dystrophy and the specific medical problems present. Myotonic dystrophy is a progressive disease, meaning that symptoms worsen as a person gets older.
- Although evidence is limited, life expectancy appears to be reduced for people with myotonic dystrophy type 1 (DM1). The most common causes of death in people with DM1 are respiratory and cardiac (heart) symptoms. An increased risk of death may be associated with younger age of onset, more severe muscle weakness, and cardiac conduction defects. People with more mild symptoms of DM1 may have a normal lifespan.
- Definitive information about prognosis in people with myotonic dystrophy type 2 is limited, but the condition generally runs a milder course. People with myotonic dystrophy type 2 may have a normal lifespan. While mobility may be impaired at an early age, the ability to walk is often retained until around 60-years-old.
2) Myotonia congenita:
- (Congenital myotonia) of which two types called Becker’s disease and Thomsen’s disease exist. Both diseases are caused by mutations in the gene CLCN1 encoding the ClC-1 ion channel. More than 130 different mutations exist in total, and a large phenotypic variation is therefore present in this disease. The mutations are loss-of-function mutations that render the ClC-1 ion channel dysfunctional to varying degrees, with reduced chloride conductance as a result. Reduced chloride conductance may result in myotonia, due to the accumulation of potassium in the transverse tubules in skeletal muscle (see myotonia congenita). This is the same genetic disease that makes certain strains of North American goats faint when scared.[citation needed]
- Symptoms of myotonia (documented in myotonia congenita) are more frequently experienced in women during pregnancy.
- Myotonia could be caused by genetic mutations in the SCN4A gene that encodes the skeletal muscle sodium channel subtype 4. Some studies have suggested that changes in physiological pH could have modulatory effects on Nav1.4 sodium channels, which could have manifestations in myotonic phenotypes.
3) Paramyotonia congenita:
- This disease results from a mutation in the SCN4A gene encoding the voltage-gated sodium channel Nav1.4 in the skeletal muscle fiber membrane. Mutations may alter the kinetics of the channel, such that the channel fails to inactivate properly, thus allowing spontaneous action potentials to occur after the voluntary activity has terminated, prolonging relaxation of the muscle, or can result in paralysis if the relaxation is severely prolonged. This inability of muscles to relax worsening with exercise is often termed “paradoxical myotonia.” Paramyotonia also frequently triggered by exercise, cold, and potassium.
3) paramyotonia congenital:
- This disease results from a mutation in the SCN4A gene encoding the voltage-gated sodium channel Nav1.4 in the skeletal muscle fiber membrane. Mutations may alter the kinetics of the channel, such that the channel fails to inactivate properly, thus allowing spontaneous action potentials to occur after the voluntary activity has terminated, prolonging relaxation of the muscle, or can result in paralysis if the relaxation is severely prolonged. This inability of muscles to relax worsening with exercise is often termed “paradoxical myotonia.” Paramyotonia also frequently triggered by exercise, cold, and potassium.
4) Potassium-aggravated myotonia:
- Potassium-aggravated myotonia (PAM) results from in a mutation of the SCN4A gene that causes skeletal muscles to be unable to relax after contracting in bouts, typically following the consumption of potassium-rich food.[14] It is debated if potassium-aggravated myotonia is a distinct disease from Paramyotonia Congenital, and recent academic papers have classified it both ways.
5) Hyperkalemic periodic paralysis:
- Also known as HyperKPP. Similar to Paramyotonia Congenita, where potassium exacerbates myotonia in many phenotypes, Hyperkalemic Periodic Paralysis is another disorder of the SCN4A gene where high blood potassium levels result in muscle weakness, muscle paralysis (through weakness or through overexcitation preventing movement), and sometimes myotonia. Many phenotypes of HyperKPP result in issues regulating blood potassium levels, often cause it to be high or causing hyperkalemia, further exacerbating the condition.
6) Hypokalemic periodic paralysis:
- Also known as HypoKPP. Similar to HyperKPP above, except that it’s triggered by (and often causes) low potassium levels and hypokalemia. It too can result in myotonia, in addition to weakness and paralysis (from both lack of and excess signal to muscles). It also has been found to occur due to gene mutations other than SCN4A.
7) Neuromyotonia:
- Neuromyotonia (also known as Isaac’s Syndrome or NMT) causes peripheral nerve hyperexcitability that causes spontaneous musNeuromyotonia (also known as Isaac’s Syndrome or NMT) causes peripheral nerve hyperexcitability that causes spontaneous muscular activity resulting from repetitive motor unit action potentials of peripheral origin. 100-200 cases have been reported. cular activity resulting from repetitive motor unit action potentials of peripheral origin. 100-200 cases have been reported.
8) Other:
- Myotonia occurs also in certain types of limb-girdle muscular dystrophies, myofibrillary myopathies, distal myopathies, and inclusion body myopathies. Other channelopathies may cause it as well. It is also associated with Schwartz–Jampel syndrome.
Physiotherapy treatment:
Physiotherapy will aim to:
- Maximise physical potential.
- Maximise respiratory function.
- Prevent soft tissue shortening.
- Maximise motor development and milestones.
- Maintain muscle strength in order to delay muscle wastage.
- Improve postural stability.
- Improve independence.
- Improve quality of life.
Physiotherapy for babies with myotonic dystrophy
Physiotherapy treatment for babies with myotonic dystrophy will commence shortly after birth and will be centred around:
- Improving breathing to maximise lung function.
- Helping jaw and lip weakness.
- Encouraging movement.
- Facilitating postural stability such as learning how to position your child in lying and sitting to encourage. appropriate postural responses.
- If you child has club foot, gentle passive stretching will help realign the foot and ankle and prevent deformities. This may be used in combination with plaster casting.
Physiotherapy for young children with myotonic dystrophy
Physiotherapy treatment will help young children reach their developmental milestones such as physical achievement with sitting unaided, moving their head, crawling and walking. Physiotherapy treatment will target these milestones and any other difficulties with physical function and may include:
- Passive and active stretching to prevent soft tissue shortening.
- Exercises in a supported and stimulating environment to improve muscle strength and flexibility. Using toys and mirror imaging can help keep your child motivated.
- Activities to improve balance and coordination.
- Exercises to improve posture in lying, sitting and standing.
- Advice about supportive seating to prevent postural deformity and allow more efficient upper limb function such as gripping and writing.
- Advice about orthotics to improve standing and walking.
Physiotherapy for adults with myotonic dystrophy
- Strengthening exercises especially for the lower limb to improve mobility and gait pattern. The dorsiflexor muscles are also targeted to help lift the foot up when walking.
- Exercises to improve balance.
- Breathing exercise to reduce the risk of chest infections.
- Hydrotherapy to stretch tight muscles and strengthen muscles.
- Advice on orthoses to reduce the impact of foot drop and improve stability when walking.
- Advice on mobility aids such as walking sticks and wheelchairs.