Myotonic Muscular Dystrophy (DM)
Table of Contents
What is Myotonic Muscular Dystrophy?
Myotonic dystrophy is an inherited sort of genetic disease that affects the muscles and other body systems. folks that have Steinert’s disease have muscle wasting and weakness in their lower legs, hands, neck, and face that exasperate over time. Signs and symptoms of dystrophy usually develop when someone is in his or her twenties or thirties. The severity of Steinert’s disease varies widely among people who have it, even among relations.
The weakness and muscle wasting that happens slowly reach the purpose of disability. Usually, disability doesn’t become severe until fifteen to twenty years after the symptoms appear. The progression of muscle weakness is slower and is a smaller amount serious in those that are older when the muscle weakness is first noticed.
Types of Myotonic Muscular Dystrophy
Myotonic dystrophy (DM) consists of two major types — DM1 and DM2
Both are caused by genetic deficiencies. They lead to multisystem disorders characterized by muscular weakness and myotonia (difficulty relaxing muscles after use), cardiac abnormalities, cataracts, and other abnormalities.
DM1, the foremost common type, results from an abnormal DNA expansion within the DMPK gene on chromosome 19. DM2 occurs from an abnormal enlargement of DNA within the ZNF9 gene on chromosome 3. Within DM1 are additional subtypes, looking at a person’s age at the onset of symptoms. The age of onset is roughly correlated with the dimensions of the disease’s associated DNA expansion; larger expansions are related to earlier disease onset.
DM1 subtypes include:
Congenital-onset DM1 — may present before birth with decreased motion and is characterized by severe muscle weakness, profound hypotonia, poor feeding, joint contractures, cognitive impairment, respiratory failure, and other developmental abnormalities.
Childhood- or juvenile-onset DM1 — starts during childhood (after birth but before adulthood) and is characterized by cognitive and behavioral symptoms, muscle weakness, myotonia, anxiety, mood disease, attentional defaults, and other symptoms. Some patients may have arrhythmias when playing sports, and in 10% of patients, cardiomyopathy and coronary failure may well be diagnosed.
Adult-onset DM1 — begins in adolescence or early adulthood and is distinguished by slowly progressive cardiac abnormalities, respiratory weakness, weakness, myotonia, cataracts, and, sometimes, mild to moderate cognitive difficulties.
Mild DM1 – usually seems in 20- to 70-year-old patients, usually after age 40. This gentle type of DM1 is distinguished by mild weakness, myotonia, and myopia.
DM2 —
sometimes called proximal myotonic myopathy ( PROM) — has not been seen in a very congenital-onset form and irregularly begins in childhood. Therefore, it’s not described in subtypes. DM2 tends to involve the proximal muscles (close to the middle of the body) instead of the distal muscles (far from the middle of the body) that are the primary to be affected in DM1. In general, DM2 could be a less severe disease than classic DM1. However, it’s going to affect walking ability before DM1 because it causes early weakening of the hip muscles. DM2 is rare in comparison to DM1, except in people of German descent.
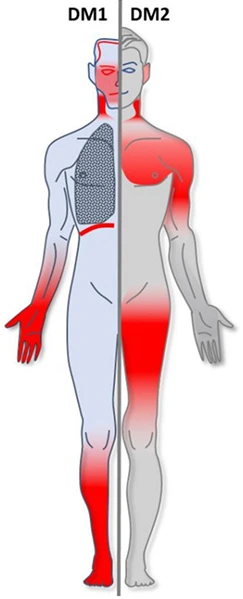
Below could be a general comparison of the foremost features of DM1 and DM2.
Comparison of DM1 and DM2.
| Feature | DM1 | DM2 |
|---|---|---|
| Age of onset | Birth to adulthood | 10 to 60 years |
| Facial weakness | Prominent | Mild |
| Neck muscle weakness | Common, early | Common, early, less severe |
| Hip and thigh weakness | As a result of a gradual progression | Early, presenting feature of DM2 |
| Distal muscle weakness | Prominent | Proximal weakness |
| Muscle pain | Common symptom | Induced by exercise and temperature change |
| Myotonia | Occurs | Occurs |
| Early cataracts (clouding of the lens in the eyes) | Occurs | Occurs |
| Cardiac rhythm abnormalities | Common | Variable |
| Hypersomnia is excessive daytime sleepiness (EDS) | Common | Less prevalent and less severe3 |
| Cognitive impairment | Occurs often; can be mild to severe | Can occur, generally mild, and intellectual disability is rare |
| Respiratory abnormalities | Common, particularly sleep-disordered breathing and inadequate breathing | Rare |
| Gastrointestinal disturbances | Difficulty to swallow, abdominal pain, diarrhea, constipation, irritable bowel syndrome, bloating, and gallstones can occur | Mild difficulty swallowing solid food, abdominal pain, and constipation |
| Uterine dysfunction during labor and delivery | Can occur | Not common |
| High blood sugar because of insulin resistance, diabetes | Can occur | Can occur; the prevalence of diabetes is greater than in DM1 |
How serious is DM?
MMD varies widely from person to person, even within the same family.
Some people don’t have any symptoms, or the symptoms are so mild the person doesn’t know they need the disease. they could learn they need MMD given that they need genetic testing after a relative is diagnosed.
In people, the symptoms are easier to note. They aggravate over time, usually slowly. MMD may change your child’s abilities and result in a variety of health concerns, from mild to serious.
The most severe MMD, which starts at birth or soon after (congenital MMD), is often life-threatening. Newborns with symptoms that will be caused by MMD typically need neonatal medical aid.
Myotonic muscular dystrophy at Seattle Children’s
Seattle Children’s Neuromuscular Program offers the foremost comprehensive care within the geographic region for youngsters with dystrophy.
The experts you wish are here
- We have been named an MDA Care Center by the hereditary disease Association.
- Our dedicated team – doctors, nurse practitioners, therapists, dietitians, genetic counselors, and social workers – has expertise and knowledge in diagnosing and treating MMD. Many healthcare providers within the community aren’t as acquainted with MMD because they rarely see children who have it.
- Seattle Children’s brings together pediatric specialists from Rehabilitation Medicine, Neurology, Pulmonary and Sleep Medicine, and also the Heart Center to judge your child’s unique needs.
Treatment custom-made for your child
- We design and supply care to assist your child and family manage MMD and to relinquish your child the most straightforward possible function and quality of life now and as they grow.
- Seattle Children’s provides a full range of treatments, from medicines, therapy, and movement aids to mechanical ventilation, heart care, and orthopedic surgery.
We take a team approach
- In the Neuromuscular Program, your child may even see their entire team in 1 room on one day, making the method easier on you and your child.
- To make sure your child encompasses a full evaluation and receives complete, coordinated care, we are going to involve experts from across Seattle Children’s.
- Based on your child’s needs, may include experts from Orthopedics, Orthotics, Speech and Language Services, Neurodevelopmental Clinic, and other specialties and programs.
What is the outlook for youngsters with DM?
Each child with MMD is exclusive. It’s difficult for doctors to predict how MMD will affect children. In general, the older your child is when symptoms begin, the milder the results and therefore the more slowly the disease worsens.
MMD doesn’t always cause serious disability, but it can. Usually, children with more serious inability start having symptoms in babyhood.
There is no cure yet for this disease, but treatment to house symptoms and forestall or manage complications can make a giant difference in your child’s quality of life. Together, you and your child’s healthcare team can set goals and make careful choices to assist your child to learn and grow to their fullest.
Epidemiology
The prevalence of DM1 ranges from 5 to twenty per 100,000 (1:20,000–1:5000). Up to 48 per 100,000 (1:2100) people tested positive for the mutation of DM1 in the big apple, although not all of those individuals would become symptomatic. Again in the big apple, permutations for DM1 were found in 191 per 100,000 (1:525). DM2 prevalence isn’t known, but genetic studies estimate it to be as high as 1:1830. DM affects males and females approximately equally. About 30,000 people within u. s. are affected.
However, recent studies suggest that type 2 is also as common as type 1 among people in Germany and Finland. The prevalence is also as high as 1 in 500 in regions like Quebec, possibly thanks to the founder effect. The incidence of congenital myotonia atrophica is assumed to be about 1:20,000.
Causes of Myotonic Muscular Dystrophy
Causes/Inheritance
What causes DM?
Type 1 Steinert’s disease (DM1) and sort 2 Steinert’s disease (DM2) are both caused by abnormally expanded stretches of DNA. The expansions occur in two different genes but appear to own similar effects on various cells, particularly the cells of the voluntary and involuntary muscles, including the guts and system. DM provides an example of a mechanism of the disease called RNA toxicity, which ends up from the expanded repeats within the flawed gene transcripts. Also, it’s known that the repeat expansions exert a dominant toxic effect on other genes not localized to either the DM1 or DM2 genes, which is understood as a “trans” effect.
DNA expansion in the DMPK gene causes DM1
In DM1, the abnormal DNA expansion is within the DMPK (dystrophia myotonica protein kinase) gene on chromosome 19 q 13.3. The defect was identified in 1992 because of the explanation for DM1.
The DNA building blocks cytosine, thymine, and guanine (abbreviated as CTG) are repeated more times than average during this disorder. the traditional number of “CTG repeats” within the DMPK gene is fewer than 35 repeats.
In DM1, there are often hundreds or perhaps thousands of CTG repeats within the DMPK gene. In DM1, the quantity of repeats correlates with the age of onset and therefore the severity of the disorder. However, in DM2 there’s no definite correlation between repeat length and also the severity of the disease. it’s important to recollect that these correlations are by no means perfect and will not be taken as absolute predictors of the course of the disease.
DNA expansion in the ZNF9 gene causes DM2
The underlying reason behind DM2 was identified in 2001 as an expanded DNA section within the ZNF9 (zinc finger 9) gene, also called the CNBP gene, on chromosome 3q 21.3.
In this disorder, the expansion contains four DNA building blocks — two cytosine molecules followed by thymine and guanine (abbreviated as CCTG) — repeated way more times than average. the conventional gene has 11 to 26 repeats; on genes of these with DM2, there are from 75 to quite 11,000 repeats, with a mean of 5,000 repeats. CCTG repeat tracts also display somatic instability.
As in DM1, the results of the ZNF9 gene abnormality appear to be widespread, affecting many cellular processes. However, the correlation between repeat length and disease severity or age of onset isn’t clear in DM2.
DM1 and DM2 are dominantly inherited
Both DM1 and DM2 are inherited in an autosomal dominant pattern, meaning it takes just one flawed allele, one copy carrying the abnormal expansion, to cause symptoms of the disease. If one parent has the disorder, every child of that person incorporates a 50% chance of inheriting the gene flaw that causes it.
If either the sort 1 (DMPK) or the kind 2 (ZNF9) disease is passed on, the kid will almost certainly develop the disease. DM1 symptoms fairly often are milder within the parent than within the child. In DM2, this increase in severity between generations doesn’t seem to occur, a minimum of most of the time.
Genetic testing for the expanded DNA that ends up in either form of DM is often performed in several laboratories.
Genetics
Myotonic dystrophy is inherited in an autosomal dominant pattern.
Myotonic dystrophy may be a genetic status that’s inherited in an autosomal dominant standard, meaning each child of an affected individual incorporates a 50% chance of inheriting the disease. The mutation involves satellite DNA, which is tandemly repeated sequences of DNA that don’t code for a protein. The replays involved in myotonia atrophica are either 3 or 4 nucleotides long, categorized as microsatellites, and this disorder results from an abnormally rise number of those microsatellites termed microsatellite expansion.
DM1
The microsatellite expansion accountable for DM1 is of cytosine-thymine-guanine (CTG) triplet repeats, termed trinucleotide repeat expansion, and classifying DM1 as one of several trinucleotide repeat disorders. This expansion occurs at the top of the DMPK gene, within the 3′ untranslated region. DMPK is found on the long arm of chromosome 19. DMPK codes for myotonic muscular dystrophy protein kinase, a protein expressed predominantly in muscle.
Between 5 and 37 repeats are taken into account normally; between 38 and 49 repeats are taken into account pre-mutation, and although not producing symptoms, children can have further repeat expansion and symptomatic disease greater than 50 repeats almost invariably is symptomatic, with some noted exceptions. Longer repeats are usually related to the earlier onset and more severe disease.
DMPK alleles with greater than 37 repeats are unstable and extra trinucleotide repeats could also be inserted during the biological process of mitosis and meiosis. Consequently, the youngsters of people with permutations or mutations inherit DMPK alleles that are longer than their parents and thus are more likely to be affected or display an earlier onset and greater severity of the condition, a phenomenon referred to as anticipation. Repeat expansion is mostly considered to be a consequence of the incorporation of additional bases as a result of strand slippage during either DNA replication or DNA repair synthesis.
Misalignments occurring during homologous recombinational repair, double-helical brake repair, or during other DNA repair courses likely provide trinucleotide repeat expansions in DM1. Paternal transmission of the congenital form is rare (13%), possibly thanks to selection pressures against sperm with expanded repeats, but juvenile or adult-onset is equally transmitted from either parent. Anticipation inclines to be less severe than in cases of maternal heritage.
The RNA from the increased trinucleotide repeat region types intranucleoplasmic hairpin loops to the comprehensive hydrogen bonding between C-G base pairs, and it’s been demonstrated that this sequester the splicing regulator MBNL1 to create distinctive foci.
A severe kind of DM1, congenital myotonia atrophica, may appear in newborns of mothers who have DM. Congenital myotonic muscular dystrophy also can be inherited via the paternal gene, although it’s said to be relatively rare. Congenital means the condition is present from birth.
DM2
The microsatellite expansion liable for DM2 is of cytosine-cytosine-thymine-guanine (CCTG) repeats, classifying it as a tetranucleotide repeat disorder. This expansion occurs within the first intron CNBP gene on chromosome 3.
The repeat expansion for DM2 is way larger than for DM1, starting from 75 to over 11,000 repeats. Like DM1, the dimensions of the microsatellite repeat array lengthen from generation to generation. Unlike DM1, anticipation doesn’t result, because the degree of repeat expansion beyond 75 repeats doesn’t affect the age of onset or disease severity.
Signs and symptoms of Myotonic Muscular Dystrophy
Symptoms of MMD can start at any age and range from mild to severe. Your child might need some MMD symptoms and not others, and they might start having new symptoms later in life.
Effects on the brain
Research suggests that, in DM1, there are also abnormalities within the parts of the brain that determine the rhythm of sleeping and waking, making excessive daytime sleepiness a barrier to full involvement in work, school, or social life for several adults with the disorder. In some people, there’s a sort of overall “apathy” that will ensue to changes within the brain associated with DM1. Also, in patients with DM1, cognitive skills are diminished, and therefore the IQ has been shown to be lower with younger age of onset. In both classic DM1 and DM2, lobe cognitive impairment (attention deficit) worsens over time but doesn’t touch other areas of cognition. Thus, cognitive problems don’t show the identical degree of decay over time that’s typical of muscle dysfunction in DM1.
Although not the maximum amount is thought about the consequences of DM2 on personality, cognition, and sleepiness like DM1, it appears that individuals with DM2 can have a number of identical difficulties in these areas but to a lesser degree. Intellectual disability is rare in DM2.
Breathing and swallowing muscle weakness
Respiratory muscle weakness doesn’t appear to be a standard feature of DM2. However, in DM1, respiratory muscle weakness can affect the function of the lungs and divest the body of needed oxygen. Weakness of the diaphragm and other breathing muscles can cause problems getting enough oxygen when an individual is asleep, whether or not they are doing not have any symptoms of breathing difficulty while awake. Thus, respiratory problems in DM1 can cause a condition like sleep disorder, during which people stop breathing for several seconds or longer over and over an evening while asleep.
Swallowing muscles, if weakened, can result in choking or “swallowing the incorrect way” (called aspiration), with food or liquid taking place from the trachea (windpipe) to the lungs rather than down the esophagus to the stomach. Swallowing is partially voluntary and partially involuntary, and both voluntary and involuntary muscles are affected.
Cataracts
Cloudy areas of the lens of the attention that eventually can interfere with a vision — are extremely common in both DM1 and DM2. they typically occur before typical age-associated cataracts seen in people without DM.
Cataracts are caused by a chemical process within the lens, which gradually goes from clear to cloudy the way the clear white of an egg becomes opaque when cooked. the precise reason why cataracts occur in DM isn’t known.
People with cataracts may notice their vision becomes blurry, hazy, or dim, which worsens gradually over time. It often happens in both eyes, but not necessarily at an identical time or at an identical rate.

Head, neck, and face muscle weakness
The muscles of the neck, jaw, and parts of the top and face may weaken, especially in DM1. Facial weakness is a smaller amount common and milder in DM2. Wasting of the sternocleidomastoid muscles within the neck are common in DM1 and typically absent in DM2. A “dropped head posture” is occasionally encountered. In men, early balding within the front part of the scalp is extremely common, adding to the distinct appearance of DM. Eyelids may droop. The chewing muscles will be affected, which makes the face look thin.
Weak neck muscles, common in both sorts of DM, can make it hard to sit down up quickly or lift one’s head straight up off a bed or couch. The stronger trunk muscles must be used for these actions.
Heart difficulties
The heart may be affected in DM1 or DM2. Oddly, because DM is usually a muscle disease, it’s not the muscle a part of the center (which pumps blood) that’s most affected but rather the part that sets the speed and rhythm of the heartbeat — the heart’s conduction system.
It is common in DM1, especially after a few years, to develop a conduction block, which could be a block within the electricity-like signal that keeps the center beating at a secure rate. This appears to occur in DM2 likewise, although there don’t seem to be as many studies on this kind of disease. Arrhythmias or Stokes-Adams syndrome may occasionally be very early manifestations of DM1, even when neuromuscular symptoms are mild or maybe unrecognized.
Fainting, near fainting, or dizzy spells are the standard symptoms of conduction block, and these should never be ignored. Such problems may be fatal. In both kinds of DM, heart muscle impairment can also occur, although it’s not as common as conduction abnormalities.
Insulin resistance
Fortunately, the general public with DM1 and DM2 don’t have diabetes, but they will develop a diabetes-like condition that’s sometimes named insulin resistance. this implies the body makes insulin (a hormone needed for the cells to require up and use sugars), except for some reason, it takes more insulin to try and do the work because the muscle tissues don’t respond normally to the same old amounts. High blood glucose may result from insulin resistance. The prevalence of diabetes is bigger in DM2 patients than in patients diagnosed with DM1.
These conditions are less common in DM2.
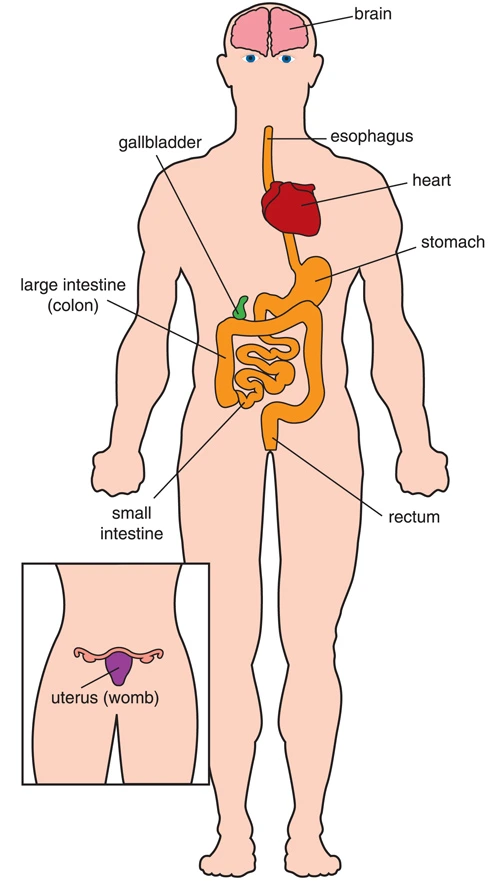
Effects on internal organs
Most of the interior organs within the body are hollow tubes (such because of the intestines) or sacs (such because of the stomach). The walls of those tubes and sacs contain involuntary muscles that squeeze the organs and move things (food, liquids, a baby during childbirth, so forth) through them. In DM1, many of the involuntary muscles that surround the crater organs can weaken. These include the muscles of the d blood vessels, digestive tube, and uterus. The digestive tube and uterus (womb) often are affected in type 1 myotonic muscular dystrophy. Also, symptoms like colicky abdominal pain, bloating, constipation, and diarrhea are common.
Abnormal action of the upper epithelial duct can impair swallowing, termed “dysphagia.” Once the food is swallowed, the involuntary muscles of the esophagus should take over and move food into the stomach. However, in DM1, these muscles can have spasms and weakness, causing a sense of food getting stuck and sometimes resulting in inhaling food into the lungs (aspiration), which might result in inhalation pneumonia. The gallbladder — a sac under the liver that compresses bile into the intestines after meals — can weaken in DM1. People with DM probably are more likely than the final population to develop gallstones. Symptoms are difficulty digesting fatty foods and pain within the upper right part of the abdomen.
These symptoms were intended uncommon in DM2, but dysphagia of solid food, abdominal pain, and constipation are reported by 41% to 62% of patients, a comparable rate to it found in patients with DM1. Dysphagia has been proved to be relatively mild, and history of bronchial pneumonia or weight loss is very uncommon. most people don’t experience incontinence or urination problems in DM.
Because of weakness and uncoordinated action of the muscle wall of the uterus, women with either style of DM may experience difficulties in childbirth that may be serious for both mother and baby. These may involve excessive bleeding or ineffective labor. Preterm labor and the risk of miscarriage are additionally more common than in women without DM. Sometimes a cesarean operation (C-section) is suggested, but surgery can also be controversial in DM.
Limb and hand muscle weakness
Weakness of the voluntary muscles usually is the most noticeable symptom for people with adult-onset DM. The explanation of DM1 is that of gradual progression in weakness.
The distal muscles (those farthest from the middle of the body) usually are the primary and sometimes the sole limb muscles affected in DM1. The areas of limbs affected may include intrinsic muscles of the hands, ankles, the forearm. The muscles that devour the foot when walking may weaken, allowing the foot to flop down and cause tripping and falling (foot drop). Falls and stumbles in patients with DM1 are 10 times more frequent than in a very group of healthy volunteers.8 Muscles of the pelvic arch, the hamstrings, and ankle plantar flexors are spared in most cases of DM1.
In DM2, proximal muscles (closer to the middle of the body) tend to point out more weakness than in DM1. Weakness within the hip girdle region is usually the presenting feature of DM2. Weakness within the upper part of the leg (thigh) occurs early in DM2. Weakness of thigh, hip flexor, and extensor muscles frequently impairs the flexibility to arise from a squat, arise from a chair, or climb stairs.
Myotonia and muscle pain
Myotonia may be a slowed relaxation following a traditional muscular contraction. Myotonia is present in all told patients with DM1, whereas myotonia is found in approximately 75% of patients with DM2. Myotonia of voluntary muscles can make it hard for somebody with DM1 or DM2 to relax their grip, especially in cold temperatures or under stress. Door handles, cups, writing by hand, and using hand tools may pose an issue, although some people never notice it. Myotonia can also affect the muscles of the tongue and jaw, causing difficulty with speech and chewing.
Myotonia is often uncomfortable and may even cause pain, although people with DM1 and DM2 can also have muscle pain that’s not connected to the myotonia. Pain is more common within the legs, where myotonia can’t be demonstrated, and is one among the symptoms (along with stiffness and fatigue) that may bring patients to medical attention before the onset of symptomatic weakness. Pain in DM2 is also induced by exercise, palpation, or temperature changes. pain may trigger a work-up for a heart condition.
Cancer susceptibility
Myotonia is related to a higher risk of cancer. specifically, significantly elevated risk (two-fold) has been reported for cancers of the endometrium, brain, ovary, and colon.
Congenital MMD symptoms
Babies with MMD symptoms after birth may have:
- Serious trouble breathing because of weak breathing muscles
- Serious muscle weakness and lack of tonus, which can make them seem floppy
- Difficulty sucking and swallowing
- Clubfoot or tight ankles
- Impaired cognitive development
- Problems with hearing, vision, and speech
- Developmental delays
- Breathing and muscle movement tend to induce better within the first number of years. Most babies learn to sit down, stand and walk. milestones might take longer to achieve than average. after they reach adolescence, children with congenital MMD start to possess symptoms of adult-onset MMD.
Childhood-onset DM symptoms
If symptoms begin during childhood but after infancy, the primary symptoms are usually:
Intellectual disability, like struggling to plan ahead, make decisions, process visual or spatial information, or listen
Learning disabilities.
Your child might also:
- Avoid communicating with people or behaving differently around people out of interest in being assessed (avoidant personality)
- Seem to lack emotion or interest (apathetic personality), which could come from trouble organizing and processing information and making decisions, instead of not caring
- Over time, A child will start having muscle difficulty and other symptoms of adult-onset MMD.
Adult-onset DM symptoms
If symptoms begin during adolescence or young adulthood, the primary thing you’ll notice is weakness in muscle that slowly gets worse.
In MMD1, weakness usually starts in:
- The neck and face may cause drooping eyelids, trouble swallowing, and a weak smile.
- The fingers, hands, and forearms, might cause a weak grip
- The feet and lower legs, which might cause foot drop
- Over time, weakness can spread to other muscle groups, just like the breathing muscles and upper legs.
health effects or other symptoms of adult-onset DM1 may include:
- Myotonia
- Shrinking muscles (wasting)
- Problems with muscles within the system, which might result in choking, getting food or liquid within the airways (aspirating), gallstones, constipation, diarrhea, or pain
- Not breathing to a tolerable degree, especially during sleep
- Arrhythmia and cardiomyopathy
- Cognitive impairment or learning disabilities
- Avoidant personality or apathetic personality
- Being unusually sleepy during the day (which may need to do with weakness, poor breathing, effects of MMD on the brain, or other factors)
- Hormone problems, like hypothyroidism, insulin resistance, diabetes, or, in males, hypogonadism that results in infertility
- Some of these effects, like cardiomyopathy and cataracts, don’t seem to be likely to affect your child for several years, if at all.
MMD can affect the muscles of the uterus, so girls and girls with DM need special care if they become pregnant.
Diagnosis of Myotonic Muscular Dystrophy
A diagnosis of Steinert’s disease could also be suspected based upon an intensive clinical evaluation, a close patient and case history, and identification of characteristic physical findings. A case history of muscle weakness and myotonia could be a strong indicator of a diagnosis for DM.
Clinical Testing and Workup
Molecular inherited testing can certify a diagnosis of DM1 or DM2. Molecular genetic testing features for changes or modifications within the DMPK gene known to cause DM1, or within the CNBP gene for DM2. However, this testing is accessible only as a diagnostic service at specialized laboratories.
Electromyography (EMG): it’s a test that records electrical activity in skeletal (voluntary) muscles at rest and through shortening. An EMG can show characteristic alterations that indicate the attendance of myotonia or myopathy. This was a routine test for DM before molecular genetic testing was developed. The EMG changes aren’t specific for DM1 or DM2.
Blood tests: Individuals with DM may have mildly or moderately elevated levels of a muscle enzyme called creatine kinase or CK in their bodily fluid. Some individuals have low levels of IgG. Immunoglobulins are specific proteins produced by certain white blood cells. They play a job in defending the body against foreign substances or microorganisms by destroying them or coating them so that they are more easily destroyed by white blood cells.
A specialized imaging technique called resonance imaging or MRI may be accustomed create images of the brain. An MRI uses a force field and radio waves to provide cross-sectional images of particular organs and bodily tissues. In DM, this may show characteristic changes within the brain including degeneration (atrophy) of the cerebellum, the world of the brain that controls movement and balance.
Liver function tests: They should show elevated levels of liver enzymes in some people. The reason behind this elevation is unknown. The liver function test abnormalities in DM1 and DM2 are also misinterpreted as an indication of hepatitis or another disease.
To understand how DM has effects on your child, your Seattle Children’s team may suggest other tests and exams, such as:
- Tests of motor skills and strength
- Sleep studies
- Vision exam
- Swallowing evaluation
- Cognitive tests
Treatment Of Myotonic Muscular Dystrophy
Treatment of Steinert’s disease is by a multidisciplinary team. A neurologist oversees the assorted needs of the patient and directs care. The neurologist may recommend that myotonia, the lack of relaxed muscles, be treated with a drug like mexiletine. An EKG to appear at the guts rhythm, and often an echocardiogram to appear inside a function, are going to be performed. A test of lung function also will be performed. betting on the neurologist’s findings and results of those tests, a referral to other Johns Hopkins specialists who even have expertise in muscular dystrophy, including cardiologists, pulmonologists, and ophthalmologists is recommended for added treatment.
Specialists in rehabilitation medicine are present during clinic time to satisfy patients and supply individualized exercise and stretching programs for the treatment of weakness and contractures. Also on an identical day, the patient is evaluated for the requirement for splints and orthotics to assist with hand or foot function. genetic disorder Association liaisons are readily available to deal with equipment needs also as social and financial issues.
medical treatments.
Medical Treatment
This section addresses the medical management of the various symptoms of adult-onset DM1 and DM2, in addition to childhood-onset DM1. These three kinds of DM share similar medical management strategies. Multidisciplinary surveillance and management of those and other issues are perfect. Suggestions regarding management are based more on the agreement and clinical experience than on demonstration from irregular controlled trials.
Anesthesia warning
An unusually high rate of complications and even deaths related to anesthesia given during surgery are reported in people with DM1. this could occur whether or not DM is mild. In fact, mild cases may be particularly dangerous because the surgeon, anesthesiologist, and patient are also less likely to concentrate on DM-associated symptoms when planning surgery. anesthesia is also appropriate for a few procedures, and for a few patients with DM1.
Surgery usually may be safely undertaken with careful monitoring of cardiac and respiratory functions before, during, and after the procedure. make certain to inform the whole medical team, especially those accountable for anesthesia, that you simply or your friend has DM. If possible, have the anesthesiologist and therefore the neurologist communicates long before the surgery.
Succinylcholine should be avoided further thanks to its potential to cause diffuse shortening.
Adverse reactions to anesthesia don’t seem as serious in DM2. However, caution is suggested.
Medicines may help treat a variety of other DM effects, including:
- Insulin resistance
- Hypothyroidism
- Hypogonadism in males
- Constipation
Who will treat the child?
The Neuromuscular Program team includes experts from Rehabilitation Medicine, Endocrinology, Neurology, Pulmonary and Sleep Medicine, and therefore the Heart Center, also as a dietitian, genetic counselor, and public servant.
Based on the child’s needs, we involve professionals from other clinics and programs around Seattle Children’s, such as:
- Gastroenterology for digestive concerns
- Neurodevelopmental to test for learning disabilities or challenges
- Speech and Language Services if the child has trouble swallowing or communicating
- Ophthalmology to gauge and treat eye problems
- Newborns with congenital MMD are treated in our Level IV neonatal medical care unit (NICU) by doctors who understand this type of disease.
Treatments for movement and performance
Common treatments to take care of your child’s ability to maneuver and do the items they require to try and do include:
- Physical therapy to take care of tonicity and improve range of motion
- Occupational therapy to assist your child with activities like dressing and employing a keypad
- Exercise and stretching to take care of your child’s strength and endurance, improve tight muscles and joints (contractures), and control pain
- Splints or braces for your child’s hands or feet to supply support so your child can do more on their own
- Medicine to alleviate myotonia or pain in the muscle
- Devices that help your child get around, like a walker or wheelchair, or that help them with daily tasks, like a special chair for sitting in the shower
- Surgery to release contractures (tight muscles and joints).
- Breathing and coughing muscle weakness
In DM1, breathing muscle weakness will be a crucial consider the disease course. It doesn’t seem common in DM2. Obstructive sleep disorder may coexist with a diminished breathing drive, especially in those at the highest risk due to coexisting obesity.
A good thanks to treat respiratory muscle weakness is to pump air into the lungs during the night with a tiny low, portable “breathing booster” called a bilevel positive airway pressure device (BiPAP is the trademarked name of the device made by Philips Respironics). it’s usually used with a mask which will be easily placed on and brought off. this type of breathing assistance can also be used during the day, although usually, that’s not necessary.
Cough assistance machines and assisted cough techniques can help people filter secretions, especially when an individual with DM1 features a cold or chest infection. An MDA Care Center physician, respiratory therapist, or pulmonologist can advise about these techniques and machines and the way to use them.
Daytime sleepiness
Daytime sleepiness, which is more common in DM1 but also occurs in DM2, sometimes can be helped with medication. Methylphenidate (Ritalin) and modafinil (Provigil) were shown to helpful and well permissible in treating excessive daytime sleepiness.
Another approach that may be tried is to coax the body into an enhanced rhythm of sleeping and waking exploring bed and getting up at the same time on a daily basis. check with a respiratory expert accustomed to dystrophy to see if breathing is undermined during sleep.
The daytime sleepiness of DM1 is also aggravated by breathing issues. Thus, it’s critical to assess the standard of sleep in patients with DM1, looking for whether there are unexplained awakenings, nocturnal restlessness, and/or loud snoring punctuated by occasional awakening and gasping for breath, all of which should suggest the presence of a sleep-related disease and result in further study.
Insulin resistance
A phenomenon referred to as insulin resistance (meaning the insulin produced by the body isn’t utilized further than it should be) can cause high blood glucose and sometimes even diabetes. Insulin resistance is common in people with DM1 and is assumed to affect approximately 20% of those with DM2. If it does become problematic, insulin or other medications that lower blood glucose will be prescribed.
Lung care
Infections can become serious if weak breathing muscles make it hard for your child to spit up phlegm. a tool called a cough-assist machine can help your child get a deeper breath and so clear their airways. A respiratory therapist sets up the machine and teaches you the way to use it.
Some babies born with congenital MMD may have to get on a machine to assist with breathing initially in our NICU until they will breathe well on their own. this might mean continuous positive airway pressure (CPAP), bilevel positive airway pressure (Bi-PAP), or a ventilator. Some people with MMD need CPAP or Bi-PAP to assist with breathing during sleep.
Cardiac abnormalities
Not everyone with DM needs treatment for heart problems, but everyone should be checked for heart health on a daily basis. Complications seem more common in DM1 than in DM2, although both sorts of DM can affect the guts.
The most common kind of heart problem in DM is an abnormal cardiac rhythm (arrhythmia) called a conduction disturbance. When a conduction disturbance is present, signals don’t move through the center in the normal way, this will be very serious, even causing extra time. Therefore, it’s imperative that individuals with DM have regular electrocardiograms (EKGs, aka ECGs).
DM patients may develop an abnormal rhythm referred to as cardiac arrhythmia, during which the highest part of the center beats extremely fast, causing turbulent blood flow that may result in clots and strokes.
Various electronic devices (pacemakers and implantable defibrillators) may be wont to treat abnormal heart rhythms. Sometimes, medications are also prescribed. These include some drugs as beta blockers and anti-arrhythmic drugs.
Sometimes, especially late within the DM disease course, the center muscle itself can weaken, causing a kind of disorder called cardiomyopathy. Medications will be prescribed to minimize the strain on the center during this disorder.
Diet and nutrition
Make sure the child gets the proper amount of calories to possess energy, maintain strength, and be as healthy and active as possible
Prevent or manage constipation, diarrhea, or gallstones
Babies born with congenital MMD may have a gastrostomy tube, nasogastric tube, intravenous (IV) feeding, or a special bottle to urge all the nutrients they have.
Support and welfare work
A public servant focuses on supporting your child and family and helps with coping with social relationships, behavior, and emotions. together with your child’s doctors, nurses and therapists, the caseworker will connect you with helpful resources at Seattle Children’s and within the community.
Physiotherapy Treatment
Goals of Exercise for Individuals with Myotonic Dystrophy
- Maximize range of motion and minimize muscular imbalances.
- Avoid and minimize disuse atrophy.
- Maximize functional abilities. It is important to train to maximize muscle ability to perform functional activities and daily mobility tasks such as walking, climbing stairs, and getting out of chairs. Training should be adapted to encompass varying strength throughout the full range of motion required for patient-identified functional goals.
- Energy Conservation. Watch for fatigue and overuse; consider that exercise may have a positive impact on fatigue over time.
- Prevent secondary injuries or repetitive injuries by utilizing good biomechanics.
Types of Exercise
Range of Motion/Stretching Exercise
Range of Motion (ROM) exercises are important in maintaining joint function and muscular balance and may play a role in reducing pain caused by muscular imbalance or tightness. One benefit of ROM/stretching is decreased muscle stiffness because chronically tense muscles have reduced circulation and less nutrition to muscles. Other benefits include reduced post-exercise soreness and pain, and improved mechanical function of muscles and joints by improving lubrication to the articular cartilage of joints requiring less energy to move through a ROM.
As muscle atrophy results in weakness, the gravitational pull may limit a person’s ability to move a body part through its entire ROM, therefore it may be important to change the position of the body part to minimize the pull of gravity. For example, people may have difficulty raising their arms up in a sitting or standing position, i.e. performing shoulder abduction in an antigravity position, but may have the ability to perform this movement when lying down in a supine position where the effects of gravity are
eliminated. Varying the position or assistance needed throughout the ROM is a form of strengthening throughout the ROM.
Individuals may also participate in ROM exercises that are more dynamic in nature. This includes yoga or Pilates-based activity that can either be done individually or in a class setting. Education regarding ROM exercise is essential to the management of the
symptoms related to the musculoskeletal system.
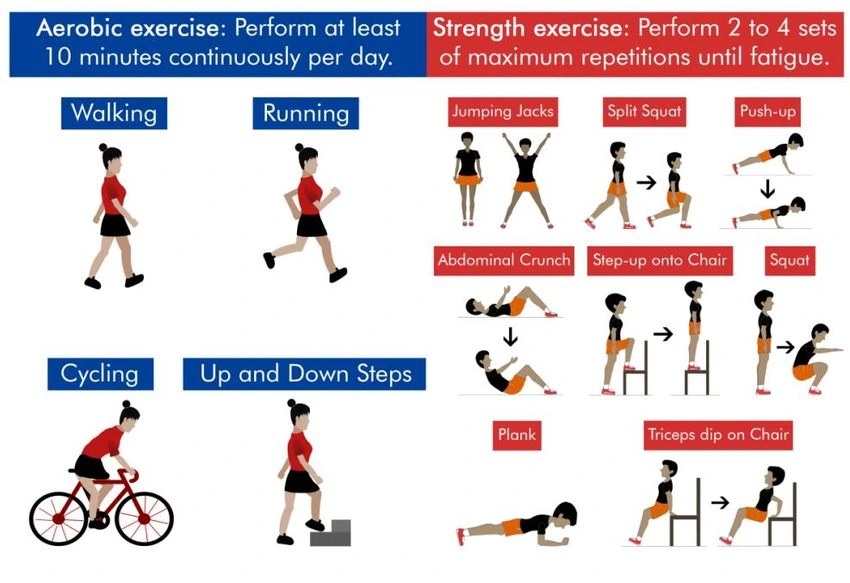
Aerobic Exercise
Aerobic (cardiovascular) exercise has been found to be safe and may offer benefits for people with myotonic dystrophy. Aerobic exercise increases your heart rate and respiratory rate and is aimed at increasing endurance and fitness. Some examples of aerobic exercise are walking briskly, running, bicycling, swimming, or exercising in water, using machines like an elliptical, dancing, or raking leaves. There are many benefits of aerobic exercise, however, because DM can affect the heart rhythm, it is essential that individuals have a physical, appropriate cardiac evaluation and clearance from their physicians prior to initiating an aerobic exercise program.
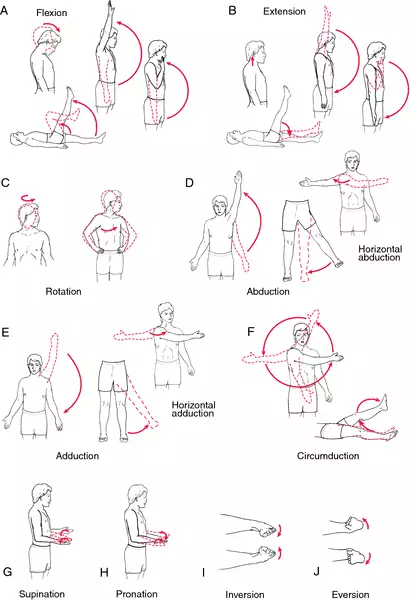
Resistive Exercise
Individuals with DM may benefit from a resistive exercise program with studies showing improvements with strength training. Resistance exercise can be accomplished in several ways with resistance provided by gravity, water (in a pool), equipment such as elastic bands, free weights, and machines. Yoga and Pilates type of exercises may be recommended as part of a strengthening program, but there are no specific studies that have examined the effects of these specific interventions in patients with DM. It is essential that individuals with DM work with providers who are knowledgeable about their condition and have a proper baseline
evaluation and appropriate follow-up to monitor and modify the program as necessary.

Balance Training
Balance training improves mobility and coordination and can reduce falls and the fear of falling. Individuals with DM tend to have fall risks similar to that of the geriatric population due to distal limb weakness and decreased reaction time. A comprehensive balance program involves maximizing sensory input, strength, and balance strategies. Individualized exercise programs require assessment and treatment from a physical therapist. However, balance training can be incorporated into daily physical activity and exercise programs. Practicing task-specific static balance and dynamic balance activities can improve balance for performing daily tasks.
One example of a static balance exercise is standing on different surfaces (hard/soft) with eyes open and eyes closed or while turning your head back and forth. Another example is balancing with feet close together, in tandem, or on one foot. These exercises can be performed near a wall or kitchen counter corners for safety. Dynamic balance exercises are activities that are performed while moving and include walking and talking or performing other mental tasks, walking and carrying objects, or more formal exercise such as yoga and tai chi. In addition to practicing balance activities, strengthening exercises that engage the trunk and hip muscles are important for stability and balance reactions and should be incorporated into an exercise program.
Finding Motivation for Participating in Physical Activity and Exercise
Participation in physical activity and exercise requires motivation. There may be individual factors such as personality, knowledge, and beliefs that are associated with adhering to an exercise program. Burnet suggests that pursuing activities that are of personal interest and fun are important factors to consider when starting a new exercise routine to improve adherence. Recently, the investigators of the OPTIMISTIC study reported that the use of Cognitive Behavioral Therapy (CBT) resulted in increased activity and reduced fatigue in individuals with DM1. Strategies such as making exercise part of a daily routine, engaging friends and family, and monitoring progress can help individuals stay motivated to adhere to exercise. Scheduling activities using calendars and alarms, and making “appointments” with friends/family members, physical therapists, personal trainers, exercise classes or other instructors can help add exercise to daily routines. Keeping a log of activities, setting goals, tracking progress, and having a reward-based system can help maintain motivation.
Monitoring Exercise
Monitoring the body’s response to exercise helps determine the level of physiological work during exercise. There are multiple methods for monitoring exercise. In an exercise lab, exercise intensity is directly measured via VO2 measurement of oxygen utilization. Other means of measuring intensity outside of the clinic include heart rate (HR), rate of perceived exertion (RPE), and talk test.
Complications
Myotonic dystrophy is often related to a spread of cardiovascular defects. the foremost common are conduction disturbances, arrhythmias, and cardiomyopathy. DM1 patients might also develop the more common age-related cardiovascular complications like artery disease and valvulopathies.
FAQ
What happens to the body with myotonic dystrophy?
It is characterized by progressive muscle weakness and wasting. People with this disorder often have prolonged muscle contractions (myotonia) and don’t seem to be able to relax certain muscles after use. for instance, an individual may have difficulty releasing their grip on a doorknob or handle.
Does dystrophy get worse?
Myotonic dystrophy is an inherited variety of inherited disorders that affects the muscles and other body systems. people that have myotonic muscular dystrophy have muscle wasting and weakness in their lower legs, hands, neck, and face that exacerbate over time.
Can Steinert’s disease be fatal?
In the most severe cases, respiratory and cardiac complications will be life-threatening even at an early age. In general, the younger a private is when symptoms first appear, the more severe symptoms are likely to be. However, the prognosis is variable because of the symptoms of this disease.
Does exercise help myotonic dystrophy?
Studies show that moderate exercise is safe and should be effective for people with myotonic muscular dystrophy. 1-4 while exercise doesn’t cure dystrophy, it can help optimize function and maintain strength.
Can you build muscle if you’ve got myotonic dystrophy?
A new study ( But only of two people) shows that it’s possible to extend muscle strength in Myotonic Dystrophy.
How is the family of an individual with myotonic muscular dystrophy affected?
Men and ladies are equally likely to pass away myotonic muscular dystrophy to their children. muscular dystrophy may be a genetic abnormality then may be inherited by the kid of an affected parent if they receive the mutation within the DNA from the parent. The disease is passed on and inherited equally by both sexes.
How fast does myotonia atrophica progress?
DM1 can develop at birth (congenital form), during childhood (juvenile form), and through adulthood (adult form). The adult form is the commonest form and typically begins during a person’s 30s. Generally, the signs and symptoms of those disorders progress slowly. this can be the foremost common type of dystrophy.